

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study











July 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Sygnature Discovery’s oncology team reflect on recent developments in oncology drug discovery presented at this year’s AACR meeting.

This year’s American Association for Cancer Research (AACR) Annual Meeting in Orlando attracted well over twenty thousand on-site attendees and over six hundred talks. Some of the Sygnature Discovery team made the trip to Florida to see what’s been happening in the oncology drug discovery ecosystem since the New Orleans meeting a year ago. Here, the team take the opportunity to reflect on some of the key topics that caught their attention…
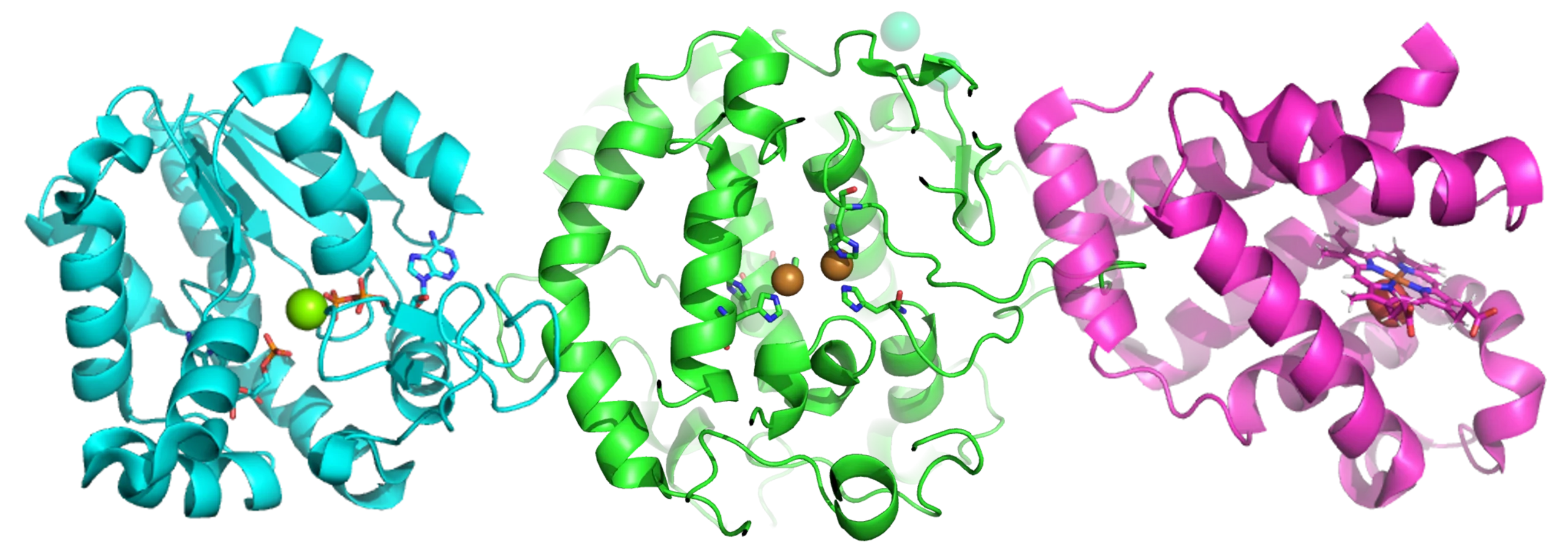
Antibody-drug conjugates (ADCs) first came to prominence back in the 1980s as a way to selectively deliver cytotoxic drugs directly to tumour cells. Comprising a tumour-specific antibody, a linker and a therapeutic payload, in theory, the antibody should target the drug to a specific tumour cell, bind to it, and become internalised. Upon internalisation, the linker is cleaved and the toxic drug is released, just where it’s needed. But it’s fair to say that ADCs have not had an easy path in the clinic. Since their inception a few decades ago, dozens of the complex molecules have entered clinical evaluation, but many have fallen by the wayside, suffering particularly from poor efficacy and systemic toxicity.
But, after a decline in interest over recent years, ADCs seem to be back with a vengeance, with several dedicated sessions at this year’s meeting. Building upon prior failures, and lessons learnt, next-gen ADCs have been engineered to improve their profiles in several important aspects. Firstly, tumour targeting has been fine-tuned, often using bispecific antibodies which bind to two different cell surface markers on tumour cells, increasing their precision. We’re also using newer techniques like digital histopathology to better understand the surface of tumours to ensure we’re actually targeting the right things on the surface of a patient’s tumour. Secondly, linker chemistries have been redeveloped, not only to improve stability (and thus decrease the release of the toxic payload in the wider systemic circulation), but also to increase the Drug-Antibody Ratio, i.e. the number of molecules of payload carried by the ADC. This higher loading, coupled with greater precision, offers the promise of a deeper therapeutic effect on tumours. Finally, this linker tuning also offers improvements in ADC pharmacokinetics, further improving the therapeutic impact and dosing convenience.
Emerging data suggests that these key improvements are now starting to deliver significantly improved therapeutic responses in the clinic. With over 160 new ADCs entering clinical evaluation, perhaps now is the time we see the promise of ADCs?
AACR always attracts some great speakers, but this year’s winner of the “AACR Award for Outstanding Achievement in Chemistry in Cancer Research” raised that bar even higher. (Though, as we sit on the selection panel for this particular award, we might be a little biased!)
Carolyn Bertozzi, winner of the 2022 Nobel Prize for Chemistry, for her role in the development of biorthogonal click chemistry, delivered an outstanding talk on her work deciphering the role played by sugars expressed on the surface of tumour cells, known as sialoglycans. These complex molecules are involved in a plethora of roles in tumour progression, in particular the interaction of the immune system with cancerous cells. Immune cells such as T-cells express families of receptors which recognise these sugar “flags”, distinguishing cells as “self” or “non-self”. By moderating how these sugars are processed, tumours can attenuate the immune response, and thus evade cell killing. Uncovering the complex regulatory biology behind this signalling is now starting to reveal novel therapeutics which may kick-start the immune system in those tumours which have modified their sialogylcans to promote immune evasion. Impressive stuff!
Equally impressive, though, was the delivery of the lecture. Dr Bertozzi decoupled the complexity of the science to ensure an engaging, entertaining and informative lecture, guiding the audience through numerous complex experiments through metaphor and analogy to ensure the participants, irrespective of background knowledge, could follow the science and the narrative. Not only a masterclass of scientific endeavour but also one of scientific storytelling and public engagement…
Amongst all the new, exciting science at AACR, it was interesting that there was a continued focus on key signalling nodes and well-validated targets. One case in point is the development of the 4th generation EGFR inhibitors, that target a range of different resistance mutations in EGFR that are acquired after prolonged treatment with 3rd generation T790M targeted therapies such as Osimertinib. It is gratifying to see that the continual learnings from the clinical application of these inhibitors are leading to new, and improved ways to prolong patient lives. It also highlights that the development of a first-in-class inhibitor versus a new target is often the start of the journey, rather than the end. Two examples of targets at the start of that journey were prominent at this year’s meeting in the shape of KRAS and TEAD.
We continue to see significant progress on directly targeting KRAS, a target once thought undruggable. The first-generation KRAS G12C-selective drugs are now approved and although patients receive significant benefits, the duration of response is limited as drug resistance emerges. The success of this approach has sparked a wide-ranging effort to develop the next generation of drugs versus this and other oncogenic forms of KRAS. Highlighted at AACR was a first look at a new small molecule that targets the “ON” state of the KRAS G12C protein; the first-generation inhibitors target the “OFF” state. These new molecules, therefore, target the most prevalent form of the KRAS G12C protein in the cell and have significant pre-clinical improvements over the first generation of inhibitors. With this molecule moving into the clinic soon, it will be of interest to see if patients receive a similar improved benefit and extended duration of responses. Of course, there are many varieties of oncogenic KRAS, and although perhaps further behind in development we are beginning to see some candidate molecules emerge that target the G12D and G13C alleles, as well as pan-mutant KRAS inhibitors. Interestingly, and perhaps inevitably, we are also starting to see the fields of KRAS inhibitors and PROTACS collide, with the first disclosure of a bi-functional degrader of the KRAS G12D mutant protein. So, although there was a justifiable celebration of 40 years of research on KRAS at this year’s meeting, it feels like the journey to drugging this target has only really begun.
This year also marked significant advances in attempts to target the HIPPO pathway. This pathway has been well studied in the context of embryonic development for many years and more recently has emerged as an important target in cancer, particularly in NF2 mutated tumours that show a dependency upon YAP/TEAD transcriptional activity. There were disclosures of several different TEAD inhibitors, the majority of which bind in an allosteric pocket and disrupt TEADs interaction with its transcriptional partner YAP. Most notably there was the first report of a phase 1 clinical trial with the first-in-class inhibitor VT3989. The drug was well tolerated and demonstrated durable anti-tumour responses. Intriguingly, these responses were observed in patients with and without NF2 mutations, potentially suggesting a broader treatable patient population than first envisaged. As these inhibitors progress in the clinic, the detective work can really begin to understand which patients will receive benefits and also to provide insights into what the next generation of TEAD inhibitor should look like.
A team from Sygnature Discovery attended this year’s American Association for Cancer Research (AACR) conference in sunny Orlando, Florida. Targeted Protein Degradation (TPD) approaches were prominent both on the clinical and pre-clinical side, breaking new ground in the battle against cancers. Here, we have summarized the TPD highlights from the conference.
Arvinas kicked off the TPD side of the meeting with a first disclosure of the ARV-766 structure, their next-generation androgen receptor (AR) PROTAC for prostate cancer, currently in Phase 1 trials. Here, a key improvement compared to ARV-110 (Bavdegalutamide) is the equipotent degradation of clinically relevant AR mutants. While trial data is still needed to understand efficacy and safety of ARV-766, it is noteworthy that preclinical in vivo dosing leads to an impressive 98% tumour growth inhibition at half the dose of the Enzalutamide (AR antagonist) reference. Alongside this, Arvinas showed data on how cancers may develop resistance to degraders (using the ER degrader Vepdegestrant), with data indicating that resistance may mainly occur as a result of upregulation of other signalling pathways (HER and MAPK), rather than loss-of-functions in the ubiquitin machinery or resistance mutations in the ER pathway.
C4 Therapeutics presented data on their BRAF V600X BiDAC degrader, CFT1946, currently in phase 1 clinical trials. A striking result here was that the change of a single atom from carbon to nitrogen in the BiDAC linker dramatically improved bioavailability from 5% up to 89%. This change increases the polarity of the compound, suggesting if anything, a reduction in predicted bioavailability. This contradiction may be explained by a behaviour called hydrophobic collapse, or chameleonicity, which is the masking of polarity by a compound folding in on itself, effectively reducing exposed polarity and aiding uptake. With excellent mouse bioavailability, CFT1946 also shows activity in a model of clinical resistance to BRAF inhibitors, highlighting the potential of degrader approaches.
On the KRAS front, Astellas showed data on ASP3082, a KRAS G12D degrader in phase 1 development. Whereas the KRAS G12C cancer mutant has been successfully targeted by exploiting covalent bond formation with the mutant cysteine (the Lumakras inhibitor), cancers driven by the G12D mutant cannot be drugged in this way. Although a compound structure has not yet been divulged, it now appears that Astellas have provided a promising approach to address this challenge, demonstrating selective and potent degradation of KRAS G12D over the normal KRAS.
Bruton’s Tyrosine Kinase (BTK), a driver of B-cell lymphomas, has been successfully drugged by Ibrutinib and subsequent inhibitors, however, resistance mutations and mechanisms potentially involving scaffold functions of BTK commonly lead to relapse in patients. NX-2127, a dual mechanism of action degrader developed by the team at Nurix Therapeutics, targets both BTK and the IKZF1/3, combining two separate anti-tumour mechanisms. NX2127 is active against multiple resistance-associated BTK mutants, and gratifyingly, could be shown to generate a complete response in a case study of a relapsed patient heavily pre-treated with Ibrutinib and Lenalidomide.
Foghorn Therapeutics showed an excellent example of this with compounds entirely specific for CPB and EP300, respectively. CBP and EP300 are highly conserved acetyltransferases, where the sequence homology in the targets has hampered development of selective inhibitors, and the data presented by Foghorn Therapeutics clearly demonstrate how deploying a degrader approach can open up for selective pharmacology.
In a different take on selectivity, Blueray Therapeutics have pursued the concept of degrader activity specifically in cancer cells, with no effect on normal cells. This is achieved by a degrader that targets a cancer-specific mutant version of the FBXW7 ubiquitin ligase. The mutant ligase is still active but carries a cysteine mutation which is recognized by a reactive compound identified in a screen, forming a covalent adduct. By using this compound to build bifunctional degraders, Blueray have made compounds that are active only in cancer cells carrying the FBXW7 mutation, and inactive in normal cells. In principle, this approach may relieve on-target toxicity and widen the therapeutic index.
One of the most interesting novel approaches in the induced-proximity field is the development of degraders that target RNA instead of protein. The general idea here is to bring a nuclease to a specific RNA using a bifunctional compound. Matt Disney (UF Scripps Institute) presented an overview of the field and recent work.
Similar to protein degraders, ribonuclease targeting chimeras (RIBOTACs) function via catalytic, event-driven pharmacology, and can offer an opportunity to target proteins that are undruggable due to a lack of ligandable pockets. A challenge here is how to robustly identify ligands for specific mRNAs, and the Disney group have made significant here, while also highlighting that many compounds that have been developed for proteins also bind to RNA, and potentially can be leveraged in RNA degrader approaches.
Not all bifunctional degraders directly recruit ubiquitin ligases – Ranok Therapeutics have built their CHAMP (chaperone-mediated protein degradation) platform around the concept of recruiting a heat-shock chaperone, HSP90, to degrade a target. HSP90 is associated with different ubiquitin ligases involved in protein quality control, and the CHAMP compounds rely on these to ubiquitinate a target protein. The high expression levels of HSP90 in many cancers, along with the potential access to multiple ubiquitin ligases are potential advantages, with in vitro data demonstrating the degradation of multiple KRAS cancer mutants.
Induced proximity can also be leveraged to abrogate protein function by sequestration, an approach explored by HALDA Therapeutics. Here, regulated Induced Proximity Targeting Chimeras (RIPTACs) drive a selected tumour-specific protein into a stable complex with a protein essential for cell viability, functionally inhibiting the essential protein specifically in tumour cells. HALDA have chosen AR as a target to develop their first compounds, with promising early data on AR RIPTACs, including an improvement in oral in vivo efficacy compared to Enzalutamide.
Due to their generally more drug-like properties, molecular glues show promise for the future of induced proximity therapeutics, and this was reflected by a large number of presentations and posters.
IMiDs (thalidomide and derivatives) form the base for all current Cereblon-based molecular glues, as well as for bifunctionals recruiting Cereblon.
Innocare have built on this scaffold to generate ICP-490, a 3rd generation oral IMiD selectively degrading the Ikaros and Aiolos transcription factors, showing significantly improved antiproliferative activity compared to previous IMiD compounds. Importantly, ICP-490 is able to overcome acquired resistance to IMiDs and is currently being evaluated in the clinic (Phase 1) for relapsed/refractory multiple myeloma. Similarly, Monte Rosa Therapeutics presented data on MRT-2359, an IMiD based degrader highly selective for GSPT1, and inactive against Ikaros/Aiolos, underlining the fact that tweaking or extending the IMiD scaffold has the potential to dramatically modify the Cereblon neo substrate specificity.
Moving away from the IMiD scaffold, PIN Therapeutics shared news on the identification of a non-IMiD Cereblon-based molecular glue degrader of CK1α, PinA1, where the compound shows anti-tumour activity in mouse xenograft models. At present, no structure has been disclosed, however, the compound is claimed to be distinct from Lenalidomide and Thalidomide, and it will be interesting to learn how PinA1 binds Cereblon, whether it is in the same pocket as the IMiDs and if this compound could also act as a handle for building bifunctional degraders. Another glue degrader that is likely a non-IMiD, pending release of the structure, was reported by Coloma Therapeutics. The compound, CL-AD-100, was identified in a high-throughput screen for degraders of ADAR1, highlighting the utility of unbiased methods for identification of novel molecular glue-type degraders.
At Sygnature Discovery, we’re passionate about delivering better therapeutics, and that’s why we work across the entire span of oncology drug discovery. By working in integrated teams made up of specialists in medicinal chemistry, DMPK, bioscience and in vivo translational oncology, we’re better able to deliver complex drug discovery projects against challenging targets, incorporating aspects such as patient alignment and engagement. And with new perspectives, different ideas and a more collaborative, integrated approach to drug discovery, the future certainly looks a lot more promising for cancer patients.
Interested in oncology? We love talking about it. Get in touch with us to discuss your project or challenge, and learn how oncology drug discovery could be made better.





Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.
