

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study







Discover precise insights into brain neurochemistry with Sygnature Discovery's in vivo microdialysis and cOFM services. With over 20 years of expertise, we design bespoke studies that reveal how compounds modulate neurotransmitter systems in health and disease. Using UHPLC/HPLC with electrochemical detection or mass spectrometry, we deliver robust PK/PD data to support confident CNS decision making.




July 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Human PK predictions are a key element of any integrated drug discovery process: if a drug is to be successful, it will need human pharmacokinetics (PK) that provide sufficient exposure for efficacy with an acceptable dose regime. So how do we do we this? And how do we help the team make decisions about which properties to optimise?
At its core, the human PK prediction of an oral drug consists of four elements:
Combined these allow us to predict the half-life of the drug; the maximal (Cmax) and minimal (Cmin) concentrations as well as the overall exposure over time (AUC, area under the curve) for a given dose.
As a project progresses, building the relationship between in vitro and in vivo data and how parameters change across pre-clinical species allows us to extrapolate to human with increasing confidence and to identify key parameters for further optimisation.
For example, if clearance can be predicted from hepatocyte incubations in rat and dog we can use human hepatocytes to predict this in man. This in vitro-in vivo extrapolation (IVIVE) technique takes into account species differences in physiology (organ weights, blood flows) and species-specific parameters for the compound such as plasma protein binding.
IVIVE is one element of physiologically based pharmacokinetics (PBPK) which can be extended to predict the volume of distribution as a function of its physicochemical properties (pKa, LogD7.4) and the predicted binding to tissue relative to plasma protein binding (PPB). As with clearance, if PBPK modelling captures the pre-clinical Vss then we can use this to predict human Vss with greater confidence.
PBPK models of the GI tract can be used to predict the extent and rate of absorption with time as a function of the physicochemical properties of the molecule (solubility, pKa, Caco-2 permeability) and how these change as the compound moves from the low pH environment of the stomach to the relatively high pH of the intestine. Solubility in biorelevant media (SGF, FaSSIF, FeSSIF) can help understand the relationship between physchem, dose and absorption and whether further form and formulation work may be needed to achieve sufficient exposure for efficacy and/or toxicology studies.
Different approaches are sometimes needed – for example, if clearance is renal rather than hepatic we need an alternative to IVIVE to predict clearance in man. Allometric relationships, where clearance and/or volume of distribution scale reliably with body weight allow us to do this. In addition, semi-mechanistic approaches such as the Oie Tozer method to predict Vss combine elements of both PBPK (species-specific physiological parameters) with measured in vivo data (Vss to allow tissue binding to be estimated). In vivo, estimates of the rate and extent of absorption can also be used to estimate the human parameters.
Ideally, the different approaches outlined give a consistent result, but they will rarely be exactly the same. An understanding of the likely variability and the impact on the prediction can be useful in understanding the risk of any uncertainty. This can be evaluated using a range of predicted parameter values or through statistical methods such as Monte Carlo simulation.

It’s important to remember that the human PK prediction is just one element of the predicted dose. To predict dose, we need to integrate the PK prediction with a suitable PK-PD hypothesis – what is the target (unbound) concentration-time profile we want for efficacy (and safety)? For example, if continuous target engagement is required, then unbound Cmin and half-life will be key parameters whereas if PD is driven by unbound Cavg then intrinsic clearance and absorption will dominate the dose prediction. A close interaction with the bioscience and in vivo pharmacology teams allows us to design studies to understand the PK-PD relationship and build this into a final human PK-PD model.
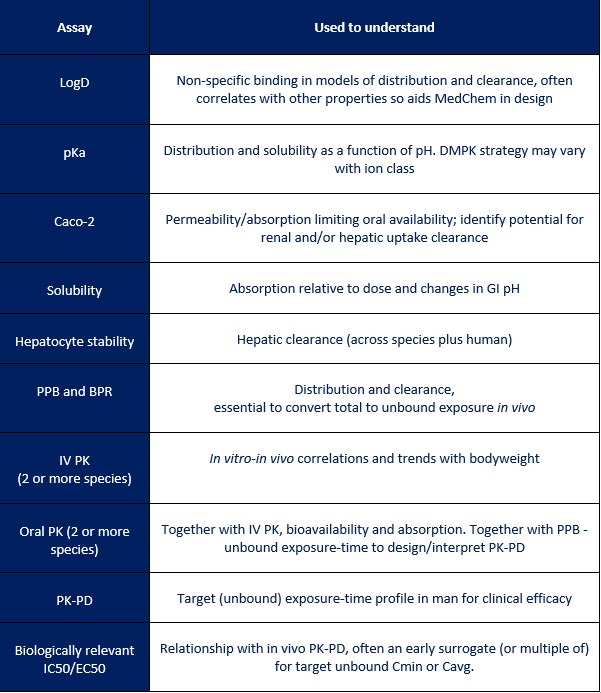
As can be seen in the table below, a lot of data is typically used in the final human dose prediction and it’s clearly not practical or economical to generate this for every compound. In reality, the PK and dose prediction evolves with time and builds in confidence as data becomes available. Working alongside DMPK in an integrated project allows the team to identify the key issues and focus on those that will really make an impact and increase the chance of success.
Graham Trevitt is the Director of DMPK Projects at Sygnature Discovery, with over 20 years of experience in DMPK. His professional background includes eight years at UCB with academic studies at Imperial College London and a PhD in Organic Chemistry from the University of Nottingham.
If you would like to find out more about how Sygnature Discovery can support your DMPK or integrated drug discovery projects, contact us today.



Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.
