

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study







Discover precise insights into brain neurochemistry with Sygnature Discovery's in vivo microdialysis and cOFM services. With over 20 years of expertise, we design bespoke studies that reveal how compounds modulate neurotransmitter systems in health and disease. Using UHPLC/HPLC with electrochemical detection or mass spectrometry, we deliver robust PK/PD data to support confident CNS decision making.




July 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
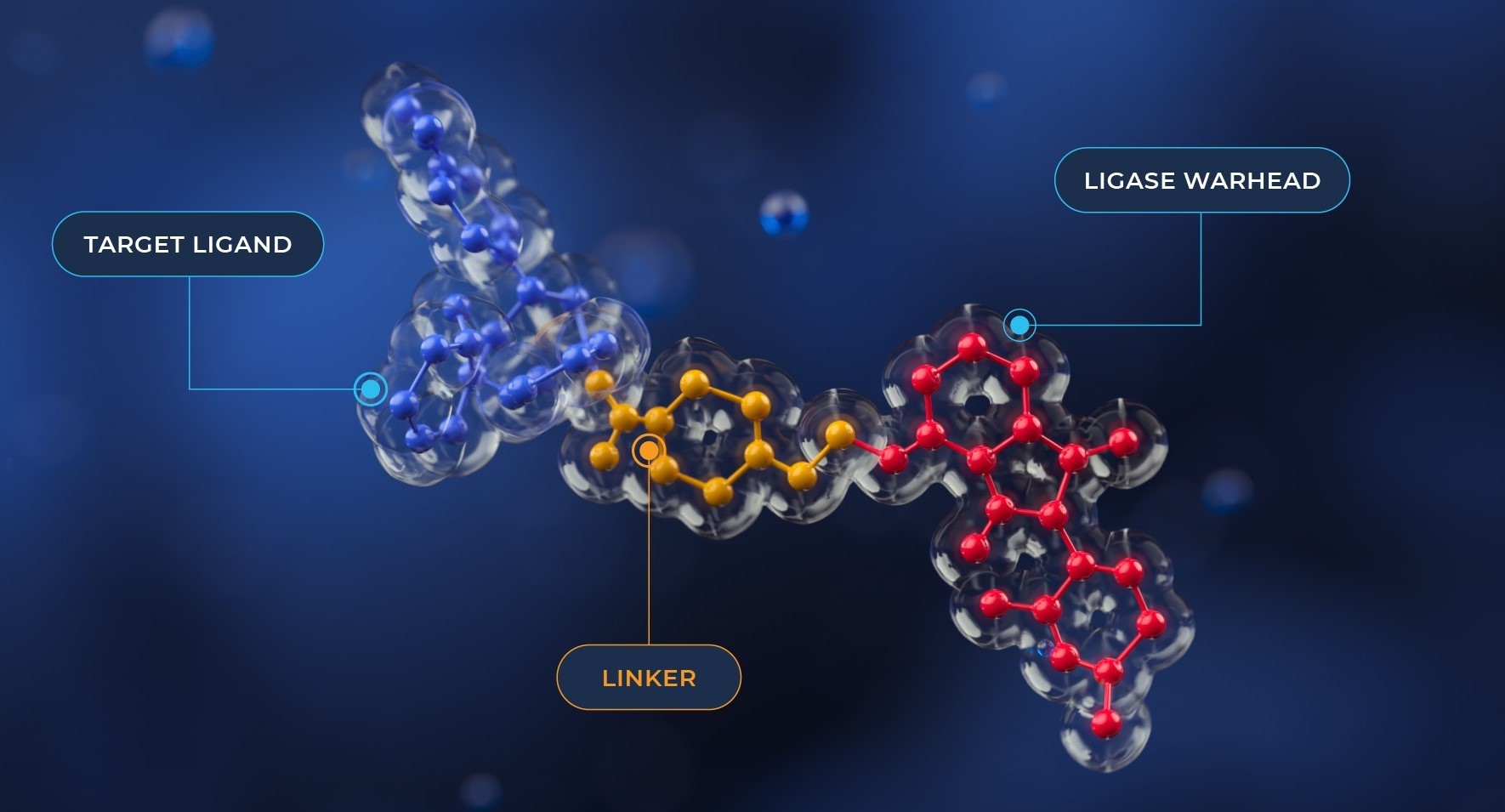
Recently, there has been a great deal of interest in the potential of PROTACs (protein-targeting chimeras) as drugs. While some are now in clinical development, it still remains early days for this therapeutic modality. Industry interest is growing, and there are various scenarios where a PROTAC could be preferable to a traditional small molecule inhibitor. This mechanism has several potential advantages over traditional mechanisms of drug activity.
Designing a small molecule that specifically binds to an active site can be difficult or even impossible in some cases. PROTACTs don’t directly inhibit a function or have to bind to active sites in the target protein. Instead, they bring the protein and ubiquitin ligase together. It doesn’t matter whether they bind to the active site, or elsewhere on the target protein. Currently, 20% of the proteome exhibits catalytic or receptor-based activity. It’s possible for PROTAC to access a significant portion of the remaining 80% of drug targets. This provides a novel opportunity to target proteins previously considered as undruggable.
Another advantage can be in achieving selectivity. The E3 ligase needs to sit in a specific geometry relative to the Protein of Interest (POI) lysine in order to achieve ubiquitination, less selective ligands could be used if they can correctly orientate the ligase with the protein of interest but not the off-target proteins. Studies demonstrate that it’s possible to achieve very specific degradation of a single protein, even if the isolated protein ligand hits multiple targets. This approach may reduce or avoid off-target effects. The benefits also lie in targeting protein families where close homology in the active site creates a challenge in achieving selectivity, for example phosphatases or certain kinase families.
PROTACs have another advantage: they can function sub-stoichiometrically. Classical drugs need to remain bound to the target over a prolonged period to have an effect, whereas once a PROTAC has mediated the degradation of a protein, it can bind to another target protein and repeat the cycle. It may take hours or even days for the protein to be resynthesized.
A single PROTAC molecule can drive the destruction of several target protein molecules, potentially reducing the required drug dose and limiting potential side effects. High concentrations of PROTACs are not required and can reduce their degradation efficiency, as shown by the “hook effect” in cells. At higher concentrations, PROTAC binary complexes start to compete with the formation of the ternary complex needed for ubiquitination.
The design of PROTACS that can induce these productive degrading complexes of the POI and the ubiquitinoylation machinery remains somewhat empirical, and currently relies on a brute force, trial and error approach. Computational simulations suggest that there likely is not a single productive complex structure. Many different orientations of the POI and the E3 ligase can lead to degradation. However, these simulations also indicate that significantly larger ensembles of unproductive conformations can occur.
Combinatorial approaches for PROTAC assembly, such as the Sygnature CHARMED platform, coupled with high throughput assessment of physical properties relevant to permeability and bioavailability, provide an effective methodology for identification of PROTAC leads. These leads have the potential to be optimized further in to drug novel candidates.





Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.
