

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study







Discover precise insights into brain neurochemistry with Sygnature Discovery's in vivo microdialysis and cOFM services. With over 20 years of expertise, we design bespoke studies that reveal how compounds modulate neurotransmitter systems in health and disease. Using UHPLC/HPLC with electrochemical detection or mass spectrometry, we deliver robust PK/PD data to support confident CNS decision making.




July 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Delve into the realm of targeted protein degradation and the critical role of formulation strategies in drug development. From overcoming dissolution limitations to optimizing stability and solubility, explore the key components that ensure your molecule’s clinical success. Sygnature Discovery offers comprehensive formulation support, bringing your degrader one step closer to achieving its full potential.
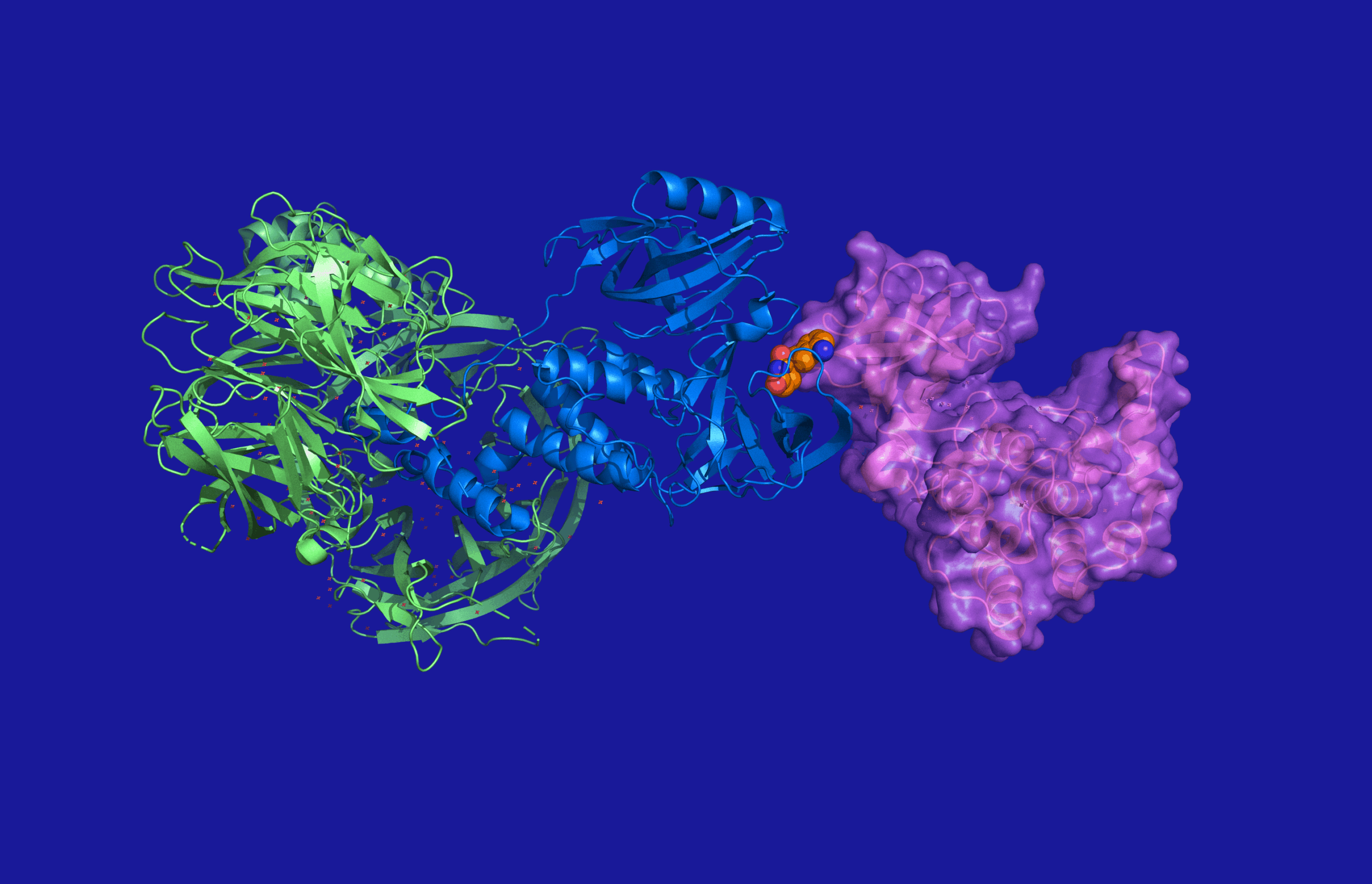
The larger the number of modalities that can deployed to treat diseases, the greater the chance of helping patients who have unmet clinical needs. An exciting new modality, targeted protein degradation, employs small molecules to induce the degradation of disease-causing proteins. Currently, most of these degrader compounds belong to the family of so-called bifunctional molecules, which consist of two moieties, a ubiquitin-ligase recruiter and a target recruiter, connected by a linker.
Many projects are in the early stages, primarily focusing on the protein to be degraded and the optimisation of linkers and ligase recruiters. Ultimately, these molecules will need to be formulated for pre-clinical and clinical testing. Degraders that are currently in clinical trials are not limited to an intravenous route of administration, exemplified by a degrader in Phase I employing an oral route of administration. In addition to dealing with the large size of these ‘small molecules’, ignoring the solid form and formulation aspect of these molecules can lead to costly delays at best, or project failure at worst.
Bifunctional degraders break several of the “rule of 5” guidelines for oral bioavailability. Early formulation strategies for the pre-clinical can enhance stability, solubility, bioavailability, permeation, and reduce efflux, offering valuable insights into the molecules’ effectiveness.
Thalidomide derivatives, known for their attractive physicochemical properties, are often used as ligase recruiters for degraders that progress to the clinic. However, this introduces a chiral centre, which is well documented to be unstable in solution. As a result, these molecules often don’t have any other stereocenters in the molecule. Removal of one of the carbonyl groups of the Thalidomide derivative renders that stereocenter stable, allowing the incorporation of stereocenters elsewhere in the molecule without fear of ending up with a complex mixture of diastereoisomers. This would make the drug substance production more complex, slow and costly, with a knock-on effect on the cost of developing the drug product.
The regulatory authorities are becoming much less reluctant to allow mixtures of optical isomers to move forward. However, the crystal landscape of such mixtures can quickly become more complex, as they could have different solubilities, dissolution profiles, and milling profiles. Any solid-state parameter could be different between these two diastereoisomers. Early solid form characterisation is recommended to identify any potential differences and mitigate any risk as you progress into formulation.
Formulating these molecules for pre-clinical species is often more challenging than that for humans. Although the use of some aggressive excipients is tolerated in the models, there may be a larger range in the amount of material required to be solubilised; this may be as little as 3 mg/ml or as high as 100 mg/ml. This is driven by the requirement of a formulation suitable for toxicology studies where dose linear PK is needed.
While most formulations for these models are in liquid form, the performance of the formation upon hitting the gastrointestinal fluids is often ignored by those unfamiliar with the nuance of drug delivery. One of the pitfalls is that the active pharmaceutical ingredient (API) can ‘dump’ out of the solution either in the gastric or intestinal fluid. Dissolution (the process of a substance forming a solution of a material) depends on various variables. These include:
A solid form screen often identifies a salt form, hydrate or polymorph which has the optimum characteristics that you are screening for. If you are unable to resolve your dissolution issues by identifying a suitable solid form, there are tools in the formulation arsenal which can be used.
Under the Developability Classification System, Class IIa compounds are limited by dissolution. The main formulation technique for enhancing dissolution is to reduce particle size. This increases the surface area to volume ratio of a particle and increases its ability to dissolve. Particle reduction can be achieved by micronisation and nano-milling.
Sygnature Discovery is well-positioned to advance your molecule toward clinical success. We possess on-site instrumentation for a wide range of formulation techniques, including lipid-based SMEDDS/SNEDDs, spray drying, lyophilization for amorphous solid dispersions, as well as simple solvent-cosolvent systems and nano-milling. We can incorporate permeation enhancers, PGP inhibitors, or combinations thereof to give your degrader the best chance of success.
To find out more about how Sygnature Discovery can support the formulation development of degraders, as well as other formulation techniques that can progress your molecule to the clinic, meet Dr Rachel Hemsley, Business Development Director, and Dr Farhan Taherali, Lead Scientist in Formulation, at CPhI Barcelona.
Contact [email protected] or [email protected] to book a meeting.





Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.
