

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study







Discover precise insights into brain neurochemistry with Sygnature Discovery's in vivo microdialysis and cOFM services. With over 20 years of expertise, we design bespoke studies that reveal how compounds modulate neurotransmitter systems in health and disease. Using UHPLC/HPLC with electrochemical detection or mass spectrometry, we deliver robust PK/PD data to support confident CNS decision making.




July 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.

Brick dust. We’ve all heard the term, but how do we solve the issue?
Poor solubility continues to be an increasing challenge in small molecule research & development1,2. The maximum in vivo exposure from an oral dose is defined by the dose administered, the form and formulation, and DMPK properties of the molecule. Unfortunately, oral bioavailability, defined as % blood exposure relative to an intravenous (IV) dose, often decreases with increasing dose.
Frustrating, right? Maybe it doesn’t have to be…
Timely engagement with the form and formulation experts could avoid the candidate molecule facing unnecessary pauses to progress.
Being successful in iterative drug discovery design requires increasing primary unbound target potency in parallel to optimising the unbound plasma/time concentration profile from an oral dose.
Typically, as the design-make-test cycle evolves, compounds are progressed to rodent pharmacokinetic studies to assess the unbound exposure, bioavailability, T½ etc. and also to understand how well this data can be predicted from DMPK and physicochemical terms (in vitro – in vivo correlation IVIV-C).
The IV (~1-3mg/kg) and oral (PO; ~5-10mg/kg) doses in PK studies are kept deliberately low for two primary reasons:
Assuming sufficient unbound exposure for efficacy is likely to be achieved, the compound will typically progress to an in vivo pharmacodynamic (PD) or disease model. Usually this will be at a higher dose range than the PK dosing unless the target potency is very high. In general, these doses can be between 3 to 100mg/kg.
To minimise animal usage in these intensive PD experiments, a dose-range-finding (DRF) study is recommended as a quick-check on dose linearity. Often this can lead to “flat-lining” of exposure whereby a 100mg/kg dose gives precious little more area under the curve (AUC) than a 30mg/kg dose as an example (Figure 1).
This creates a conundrum. What are you to do when you believe that you likely have a good compound, but no way to test it? And more so, why is this happening?
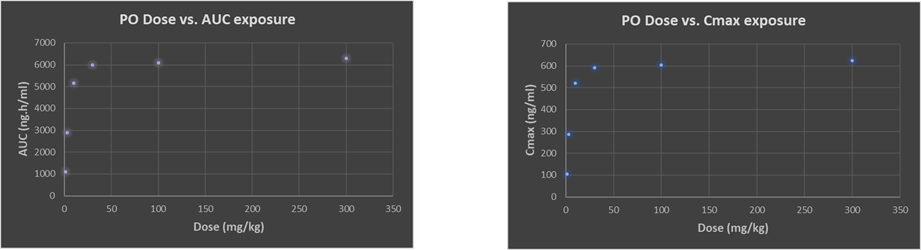
Figure 1: Typical drug discovery project example of sub-proportional plasma exposure with both AUC and Cmax as oral dose is increased
There are several theoretical reasons for this sub-proportional dose/exposure and apparent “exposure, max” ceiling. Understanding the challenges will enable us to either make improvements for this compound or design the issue out in the next round of design-make-test; but knowing where to start can feel like taking the first step up a mountain without a map.
Problems with exposure at the in vivo efficacy testing stage of the project tend to snowball as you move down the R&D timeline.
Assuming the compound is efficacious at 30mg/kg in a PD/disease model, but exposure is already showing signs of significant sub-proportionality, then demonstrating a typical suitable safety margin of 30x will require a dose of at least 900mg/kg and likely much higher due to the exposure issues.
Giving the typical upper volume of 10ml/kg on a repeat PO dose toxicity study, this will require a 90mg/ml solution or suspension.
Here you see trouble on the horizon… High concentrations ≥60mg/ml have a precedence for causing adverse effects in rodents3 and should be avoided where possible. So where do you go from here?
Assuming acceptable bioavailability (F), absorption (Fabs) and limited extraction by the gut wall (Fg) and liver (Fh) at low PK dose, but exposure flat-lines at increased dose, then contacting the form and formulation expert is the first port of call.
Working side-by-side with the in-house DMPK scientists, a collaborative discussion about the compound, its physicochemical properties, permeability, and all other DMPK understanding can pinpoint where the challenges lie.
Keeping one eye on the clinic, understanding the estimated human efficacious dose range, frequency of dosing, and dose route, will enable selection of the most appropriate solution early on, lessening future barriers to success later down the drug development path.
The scope of help is truly game-changing.
The challenge:
A Sygnature discovery partners’ drug candidate was struggling with ≤2% bioavailability with a simple suspension at moderate doses.
The action:
Following a thorough review of all the DMPK and physicochemical properties, it was understood that the compound exhibited no DMPK liabilities and the poor exposures seen were the result of the high dose requirements and low aqueous solubility in GI fluids (i.e. compound was Class IIb as per the Developability Classification System4). As the compound was neutral and salt formation was not possible, the compound was screened to identify an enabling formulation that could improve the kinetic solubility in biorelevant media and maintain high degrees of supersaturation from prolonged periods to enable more complete absorption.
The result:
In the end, an amorphous solid dispersion (ASD) was developed that gave ≥80% bioavailability and, most importantly, dose linearity from 50 to 500mg/kg.
[hubspot type=cta portal=7062255 id=a79dd693-bdae-423b-9611-a9ae6d1f8ef2]
Have we peaked your interest? Sygnature Discovery are running a two interactive webinar sessions on May 12th, and May 19th. Join us for 30 mins a time to explore how to de-risk your project, optimize the DMPK properties of your molecule, and get off the lab bench into the clinic quickly and safely with Sygnatures own drug formulation thought-leaders. View further details, access the episode abstracts, and register today here.
About the author: Dr Richard Weaver – SVP, Preclinical Development
Richard has over 25 years’ experience in the drug discovery industry from medicinal chemistry to DMPK and preclinical development. In that time he has developed an in-depth knowledge of all aspects of in vitro and in vivo DMPK, from hit identification to candidate drug nomination and beyond from his experience at AstraZeneca. After leaving AstraZeneca, Richard set up XenoGesis in 2011, which grew to be the UK’s largest independent DMPK provider, before becoming part of Sygnature Discovery in 2020.
Supporting references:





Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.
