

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study







Discover precise insights into brain neurochemistry with Sygnature Discovery's in vivo microdialysis and cOFM services. With over 20 years of expertise, we design bespoke studies that reveal how compounds modulate neurotransmitter systems in health and disease. Using UHPLC/HPLC with electrochemical detection or mass spectrometry, we deliver robust PK/PD data to support confident CNS decision making.




July 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

In drug discovery, generating targets is no longer the challenge. The real question is how to identify the few worth investing months of research and significant resources to pursue. Hear expert perspectives on how AI, pathway analysis and scientific expertise are shaping modern target identification.
July 7, 2026

Stay up to date with the latest company updates, milestones, and announcements.

Explore upcoming conferences and scientific meetings where you can connect with our team.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.

Dr. Steve St-Gallay – Principal Scientist, Computational Chemistry
The Holy Grail of computational chemistry is to accurately, precisely and routinely predict the binding potency of a potential drug molecule before it is made, and despite our best efforts (and the claims of some groups), this is not possible, yet.
The fundamental physics governing the energy between a drug molecule and its partner in the body is so complicated that it can’t be calculated without several assumptions. For example, on the atomic scale, the wonderful world of quantum physics applies, but even with access to improved computational power, these calculations cannot be performed on a reasonable timescale, so we resort to physics from the time of Sir Isaac Newton.
Another assumption is the change in disorder that occurs when a drug molecule binds to its site in the body. The universe is driven by an increase in disorder, but sometimes when a drug binds, the disorder decreases. Calculating this change in order requires knowledge of all the possible configurations, over time scales that could be as long as hours. We can only manage up to a few microseconds at best, for a single copy of the drug molecule; when you consider there are nearly 2,000,000,000,000,000,000,000 (that’s two sextillion) copies of the paracetamol molecule in a 500mg tablet, our assumptions about the change in order are probably inadequate.
Having said all of this, it does appear that with lengthy calculations we can sometimes rank the similar potential drug molecules by their interacting energy well enough to decide whether to synthesise the best ones. The figure below shows how computationally simulating the change of the molecule, both in the protein and free in water, can rank order the binding energy of potential drug compounds. The key word is ‘sometimes’; roughly a third of the time they work quite well, another third they are ok-ish, and the final third doesn’t work at all.
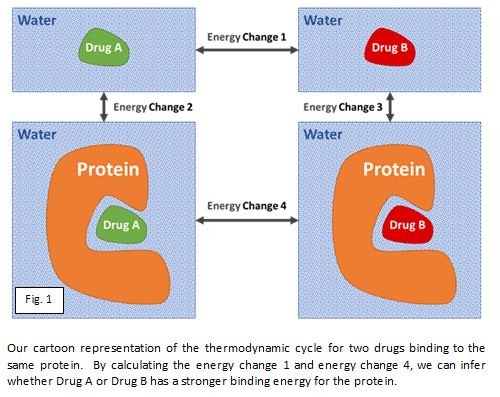
At Sygnature Discovery we have initiated a collaboration with a leading academic expert in this field to explore possible explanations for when this approach is accurate and when it breaks down, so we can then develop computational solutions to improve the accuracy.
Of particular interest is the ability of these techniques to predict scaffold hopping opportunities, since this often provides significant breakthroughs in project progress at the expense a greater synthetic chemistry investment. This will benefit the discovery and optimisation of drugs by improving the quality of compounds synthesised and reducing timelines and costs.
The search for computational chemistry’s Holy Grail continues, and perhaps we are moving a step closer with these techniques, and an understanding of the systems on which they will have the most impact.
If you would like to discuss how Sygnature Discovery can help with drug discovery resource and expertise use our ‘Get in Touch’ forms or email us via [email protected]






Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.