

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study











July 30, 2015

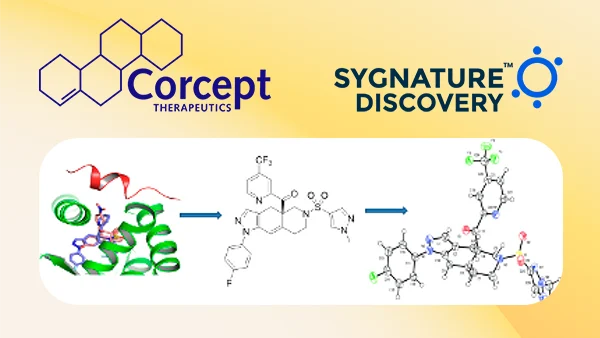
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Explore the full collection of scientific content and insight from across Sygnature Discovery.
Featured news
Blog
Sygnature scientists discuss and share their perspectives on the ideas and challenges shaping modern drug discovery.
Posters
Browse our latest posters presenting new findings, methods, and scientific advances.
Journal papers
Published research from our scientists and collaborators, highlighting contributions to cutting-edge research.
Webinars & Podcasts
Hear directly from our industry thought-leaders on topics that matter in taking drugs from ideas to the clinic.
Technical Notes
Clear, practical guidance on scientific methods, technologies, and approaches across discovery

Case Studies
Real examples of how we tackle scientific challenges and deliver impact across discovery.
Protocols
Step by step experimental protocols shared by our teams to support laboratory experiments.


Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.




























