

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study







Discover precise insights into brain neurochemistry with Sygnature Discovery's in vivo microdialysis and cOFM services. With over 20 years of expertise, we design bespoke studies that reveal how compounds modulate neurotransmitter systems in health and disease. Using UHPLC/HPLC with electrochemical detection or mass spectrometry, we deliver robust PK/PD data to support confident CNS decision making.




July 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

In drug discovery, generating targets is no longer the challenge. The real question is how to identify the few worth investing months of research and significant resources to pursue. Hear expert perspectives on how AI, pathway analysis and scientific expertise are shaping modern target identification.
July 7, 2026

Stay up to date with the latest company updates, milestones, and announcements.

Explore upcoming conferences and scientific meetings where you can connect with our team.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.

Dr Richard Weaver, Senior Vice President of Preclinical Development at Sygnature Discovery, shares his thoughts on the role of plasma protein binding in drug discovery.

It’s not just rugby scrums – binding plays an important role in drug discovery
Plasma protein binding (PPB) is an important parameter that should be measured during drug discovery and pre-clinical development in all the relevant pre-clinical species and human.
Albumin, present at a high concentration of ~4% w/v of plasma (~40 mg/ml), is the major source of binding – especially for acidic drugs – along with alpha-1-acid glycoprotein (AAG), present at a much lower 0.5 to 1.0 mg/ml in humans.
AAG is an acidic protein, and thus tends to bind basic molecules and steroids.
There are numerous oral drugs on the market and their plasma protein binding ranges from essentially 0% (Naproxen <1%, Atenolol 3%) to 99% or higher (Diazepam ~98%, Diclofenac >99%).
Perhaps this is the first clue that a specific ideal range of plasma protein binding is not required for a successful oral drug outcome.
PPB should never be used in isolation, but rather assessed along with other key in vitro and in vivo data to help compare and optimise drug discovery compounds.
This helps allow in vitro-in vivo extrapolation (IVIV-E), build pharmacokinetic (PK)/pharmacodynamic (PD) understanding and safety margin calculation, and ultimately with extrapolation from animals to humans.
“We need to optimise PPB.” “Increasing PPB will give us better PK.” “Increasing PPB will increase my half-life.” “Lower PBB equals greater free drug exposure.”
If you work in drug discovery projects, it’s not uncommon to hear these kinds of statements.
But in essence, not one of them is true – and following them as if they were set-in-stone mantras will be a futile exercise.
Let’s drill down into why and explore what the actual focus should be.
A rapid equilibrium dialysis (RED) device
Only unbound drug can contribute to wanted and unwanted pharmacology, and only unbound drug can be metabolised and excreted.
To allow the conversion from total blood or plasma exposure in vivo, you must therefore know what the plasma protein binding is.
Note that you only have to know what it is; more on that later.
At steady-state and in the absence of any transporter-mediated effects, i.e. high passive permeability across cellular membranes of the neutral form of the molecule predominates, the unbound (free) concentration in plasma equals the same unbound concentration of the molecule in tissues and the site of action.
In a practical sense, steady-state is reached after approximately five half-lives from repeat dosing (when 97% of steady-state concentration is reached).
When in vivo exposure studies are completed, total blood or plasma concentrations are measured in PK and toxicokinetics (TK).
That’s why it’s important to measure PPB in all relevant species and convert from the “total” to “free/unbound” exposure when considering PK comparisons of one compound to another, PK/PD and safety margins.
In addition, a PPB value is needed to allow a prediction of the in vivo clearance from in vitro CLint,u – thus in a typical DMPK project screening cascade, where liver metabolism is the dominant clearance mechanism, the PPB measurement should be made in parallel to the PK studies.
In other words, PPB should not be front-loaded as a key decision-making assay in a screening cascade in its own right, and should only be back-filled for compounds that progress to in vivo PK studies to allow in vitro-in vivo extrapolation and correlation.
It’s often prudent to measure human plasma protein binding at the same time to check for any differences between species, as a safety margin could easily be eroded to nothing if there are significant differences in the wrong direction.
To optimise compounds from a chemical series that are predominantly cleared by liver metabolism, arguably the most important equation to remember is:
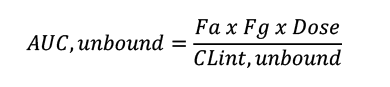
Thus, for a well-absorbed and low gut extraction compound (Fa and Fg close to 1), only dose and the unbound CLint affect AUC,unbound – with PPB not even featuring in the equation.
A very similar and useful equation for simple PK/PD planning that defines C average,unbound in plasma is:
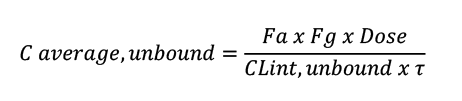
Where ???? is the dose interval.
The take-home message is that PPB should not be a target for “improvement” because it cannot be improved.
Enhancing the unbound concentration/time-profile (AUC,u) of a compound should be the primary focus, and this is achieved by decreasing the unbound intrinsic clearance of a molecule.
For a well-absorbed oral drug, this can be calculated by an intrinsic clearance experiment (CLint) in hepatocytes/microsomes followed by a conversion to CLint,u by either measuring the binding in the assay or predicting it via the multiple publications based on the lipophilicity of the compound.
It’s worth mentioning that as PPB gets very high, i.e. ≥99%, it can be very challenging to measure the binding with confidence.
Therefore, there may be a practical consideration to consider reducing the PPB in order to measure the extent of binding with accuracy and to prove whether there is a genuine difference of binding between the pre-clinical species and human.
For more detail, it’s worth reading the highly recommended, seminal scientific paper on the misconceptions of plasma protein binding written by Dennis Smith and colleagues.
Want to talk to us about how we can help your discovery project with this and other DMPK applications? Get in touch with us via the form below.





Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.