

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study







Discover precise insights into brain neurochemistry with Sygnature Discovery's in vivo microdialysis and cOFM services. With over 20 years of expertise, we design bespoke studies that reveal how compounds modulate neurotransmitter systems in health and disease. Using UHPLC/HPLC with electrochemical detection or mass spectrometry, we deliver robust PK/PD data to support confident CNS decision making.




July 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
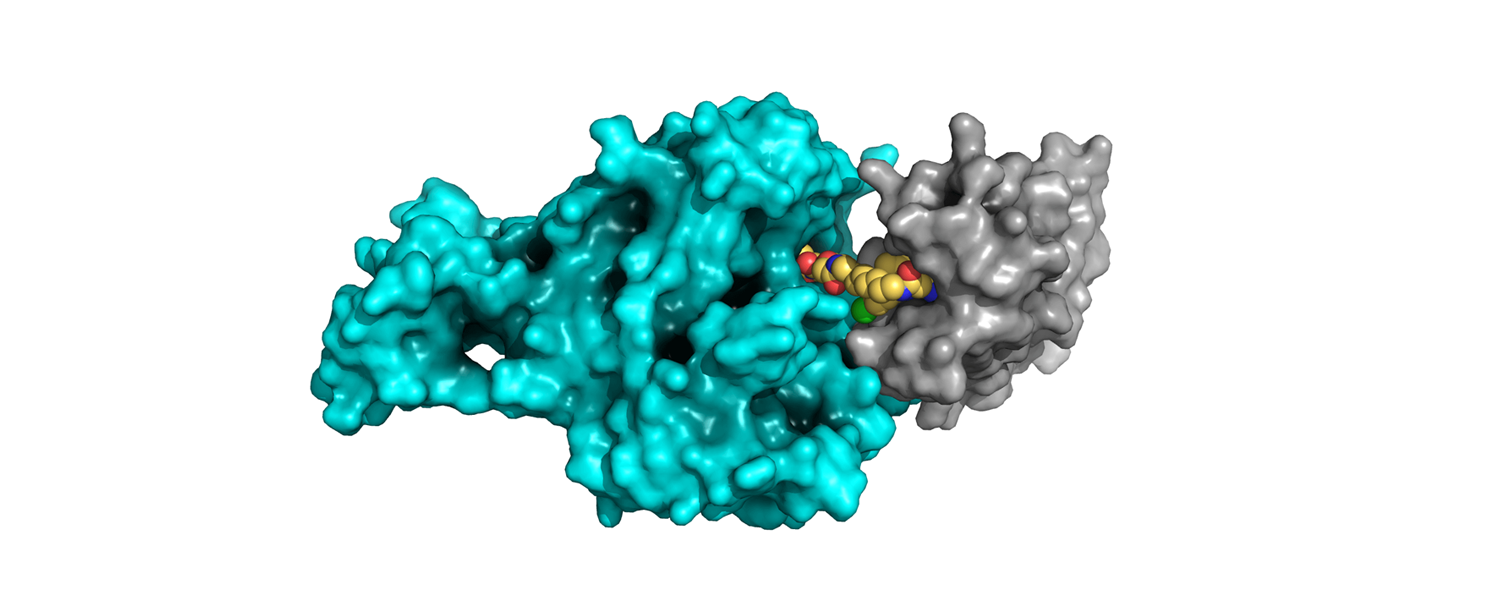
This year’s American Association for Cancer Research (AACR) Annual Meeting in April saw a welcome return to the face-to-face format. Some of the Sygnature Discovery team made the trip to New Orleans to see what’s been happening in the oncology drug discovery ecosystem since the last in-person meeting in Atlanta back in 2019. As other cancer science meetings also return to the face-to-face format, the Sygnature Discovery team have taken the opportunity to reflect on some of the key oncology drug discovery topics that have caught their attention in recent meetings…
This year’s AACR meeting coincided with the 40th anniversary of the discovery of KRAS as the first human oncogene. This was celebrated in a special session, which was chaired by Patricia LoRusso. The session included talks from Mariano Barbacid, Frank McCormack and Kevan Shokat all of whom have made significant contributions to the KRAS story. It was very interesting to hear the historical perspective, from the initial identification of KRAS as an oncogene, to the years of basic research untangling its complex biology and finally to the more recent success in drugging this, once considered “undruggable”, target.
This nicely set the scene for a wealth of presentations and posters around the various methods now being employed to inhibit this key oncogene and the signalling pathways it drives, in the clinic. The advances here perhaps point to a wider frameshift in our thinking around the concept of “undruggable”.
Given the nature of KRAS and its high affinity for GTP, approaches toward more traditional active site inhibitors were largely dismissed. Through imaginative thinking and synthetic creativity, the new realm of covalent KRAS inhibitors, targeting the key G12C mutation, have emerged, evolved and are now demonstrating clinical benefit.
However, once more, the expectation was that other KRAS family members would remain intractable. And yet, at the meeting, we saw significant advances toward truly pan-RAS inhibition, either through direct interaction or through binding partners such as SOS1 or Shp2. Seemingly, the once undruggable KRAS has now been conquered. This hard-fought success builds on the determined efforts in the early 1990s to turn kinases from an assumed undruggable protein family into pivotal first-line therapeutics for many patients. And now helicases, again heralded as undruggable, are now yielding to the efforts of Artios and others, as novel first-in-class agents approach the clinic.
As we now move toward therapeutics to interfere with transcription factors, RNA processing and other targets, alongside the active stabilisation of tumour suppressors such as p53, perhaps it is now finally time to retire the term “undruggable”? More and more, it seems, “it’s only undruggable until we learn how to do it…”
Continuing this theme of how to drug the undruggable was the increased interest in new modalities that target proteins for degradation, in particular PROTACs and molecular glues. This “recent” modality showed great promise as demonstrated by the advance of several compounds of known targets to clinical trials.
There are several advantages compared to the more “traditional” small molecules. Their potential ability to target the so-called undruggable proteome, alongside the fact that they do not need functional binding, should open the door to multiple new targets and/or hard-to-drug targets. Their catalytic mode of action might also give the opportunity of lower dosing and should, in theory, reduce the risk of side-effects and toxicity. Another possible great benefit of degraders comes from the choice of the E3 ligase, as some are restricted to particular tissues.
These efforts have significantly expanded the number of tractable drug targets as, at least in theory (although its rarely that simple!), a ligand that binds anywhere on the target protein can be used to recruit the protein degradation machinery. The PROTAC approach, using bifunctional molecules, was well represented with new disclosures on targets including Brd9 and Aurora-A.
Despite exciting progress with PROTACs, there are still some challenges that need to be overcome:
Perhaps even more exciting for the field of oncology drug discovery was the new work disclosed on molecular glues. These differ from PROTACs in being monovalent small molecules that upon binding to their target can induce a protein interface that allows recruitment of the protein degradation machinery. This is exemplified most clearly by the IMID family of molecules.
These molecular glues have traditionally been discovered in a serendipitous manner, with the phenotypic consequences of the compound treatment being observed prior to elucidation of its mode of action. At this year’s meeting, we saw a shift to a more rational approach to design these molecules.
A case in point was the development of a molecular glue targeting the transcription factor IKZF2, using the IKZF1 selective compound Pomalidomide as a starting point. An extensive structure-based design approach led to an exquisitely selective compound that was demonstrated to have anti-tumour activity in pre-clinical models through its impact on Treg biology.
A more systematic approach to discover these glues utilises cell based models, using cells mutated in a key regulatory subunit of protein degradation pathway, to allow screening of compound libraries to identify molecules that cause a phenotypic effect that is dependent upon an intact degradation machinery. This approach, and others like it, has already identified molecular glues that target Cyclin K and RBM39 and will undoubtedly lead to the discovery of new molecules in this exciting new area of chemical biology.
In an intriguing twist of this approach, we enjoyed the fascinating story on the elucidation of the mechanism of action of a compound, DNMDP, first identified in a phenotypic screen. Through extensive characterisation, the molecule was demonstrated to bind to PDE3A. However, instead of inhibiting its activity the molecule was recruiting a protein called SLFN12.
The really interesting discovery came with the realisation that as a monomer SLFN12 was a weak RNA degrading enzyme (RNase), but upon heterodimer formation with PDE3A, driven by the molecular glue, this activity was significantly enhanced. Further, this new cellular RNAase activity led to the degradation of a specific tRNA, downregulation of protein translation and ultimately cancer cell death. A wonderful demonstration of how detailed scientific investigation of mechanism of action can lead to truly novel findings that are likely translatable to other scenarios.
These presentations, along with many others in this area, herald a new wave of chemical biological approaches to uncover novel mechanisms and impact key cancer drivers. The field of oncology drug discovery can expect many more exciting discoveries ahead and, importantly, the addition of many new medicines available to our patients.
As we develop new ground-breaking medicines, getting them to patients as quickly as possible is a no brainer approach. The FDA’s Accelerated Approval programme accelerates many drugs to the clinic, where life-changing therapeutics can be approved based on early clinical data, with later clinical studies performed retrospectively.
At face value, this seems to be a hugely beneficial step forward in clinical approvals and can take years off the process by which new therapies for cancer gain approval for wider marketing and use. In doing so, access to therapeutics significantly widens, for example to patients who do not meet exclusion criteria, or who cannot access clinical trial centres, increasing inclusivity.
But perhaps all is not as it seems at first look. A recent article in Cancer Discovery (Spreafico et al., 2021, 11(4), 821) questions this approach. It argues that, counter-intuitively, accelerated approvals may actually hinder access to efficacious therapeutics. Whilst the FDA’s approval process mandates that retrospective, confirmatory trials are undertaken, these can be delayed or even not conducted. What’s more, in some cases, patients remain for significant time periods on treatments which later transpire to be ineffective in wider trials.
Indeed, only around 1 in 5 of these accelerated trials demonstrated an improvement in overall survival in larger trials, but no changes to the approval status of the other 80% of agents have been made. Whist on these trials, and potentially beyond their conclusion, cancer patients’ access to higher quality and more efficacious therapies is hindered. This potentially permits their disease to progress to a more refractory (and potentially untreatable) state.
Whilst the Accelerated Approval system helps deliver novel agents into clinical practice, reform may be needed to ensure the mandated follow-up trials are conducted in a timely and efficient manner, with robust reporting, so as not to unduly compromise overall patient benefit.
Based on the premise of “right drug, right patient, right time”, with matched therapeutics prescribed on the basis of genetic screening for known oncogenic driver mutations or fusions, this approach has been lauded as a real game changer of patients. It offers the possibility of a kinder, more effective treatment for patients with a known cancer driver.
However, even for those “fortunate” enough to have a known genetic mutation with matched therapy, the overall response rate in these trials currently stands at a woeful 7% of patients, despite enrolling over thirteen thousand patients into 37 clinical trial arms. Reasons for these abysmal figures are several-fold.
Perhaps most strikingly, alleged and identified driver mutations may not, in fact be cancer drivers. Commonly referenced oncogenes, for example PIK3CA, have recently been found not to be particularly strongly oncogenic, but sit on the same genetic amplicon as SOX2, which has been implicated as the actual drier in many PIK3CA-amplified tumours.
Similarly, many amplified FGFR1 cancers seem to be driven by co-amplification of NSD1. It seems our implied knowledge of tumour drivers may require a more rigorous analysis to ensure we are targeting the connect oncogene, alongside a better understanding of contextual tumour biology, encompassing both epigenetics and the nature of the interplay between the tumour and the immune system (reflecting the call to arms shared in our article for the British Journal of Cancer).
Further, these cancer drivers are expressed heterogeneously across established tumours, demanding the better deployment of well tolerated and thoughtful drug combinations using best-in-class inhibitors at clinically relevant dose levels. It’s also worth noting that many of these trials take place toward the end of a patient’s cancer journey, where disease is more entrenched. Perhaps enrolling patients with earlier stage disease, with higher quality agents and with a better understanding of the real tumour drivers, may eventually allow these precision medicine approaches to deliver upon their promised durable benefit?
Also under the spotlight in the clinical setting, and further reducing our ability to assess the impact of personalised therapies across the heterogeneity of cancer patients, was the issue of inclusivity and diversity in clinical trials.
Complex issues around socio-economic status, inability to access primary care, distance from a comprehensive cancer centre and a lack of accessible information dissuade or exclude significant numbers of patients from participation in trials of novel agents.
Championing the cause, and driving real change, we were inspired, enthused, and humbled to listen to the ongoing work of Prof. Robert Winn from the Massey Cancer Center in Virginia. His drive and determination in promoting community-based healthcare was infectious and is changing practices such that patients from wider racial and socio-economic backgrounds are included in clinical trials. Not only will this lead to better outcomes in often-overlooked patients, but it will significantly increase the depth, breadth and meaningfulness of data emerging from our trials.
At Sygnature Discovery, we’re passionate about delivering better drug discovery, and that’s why we work across the entire span of oncology drug discovery. By working in integrated teams made up of specialists in medicinal chemistry, DMPK, bioscience and in vivo translational oncology, we’re better able to deliver complex drug discovery projects against challenging targets, incorporating aspects such as patient alignment and engagement. And with new perspectives, different ideas and a more collaborative, integrated approach to drug discovery, the future certainly looks a lot more promising for cancer patients.
Interested in oncology? We love talking about it. Get in touch with us to discuss your project or challenge, and learn how oncology drug discovery could be made better.





Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.
