

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study







Discover precise insights into brain neurochemistry with Sygnature Discovery's in vivo microdialysis and cOFM services. With over 20 years of expertise, we design bespoke studies that reveal how compounds modulate neurotransmitter systems in health and disease. Using UHPLC/HPLC with electrochemical detection or mass spectrometry, we deliver robust PK/PD data to support confident CNS decision making.




July 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Sygnature Discovery’s Chemical Stability assay provides a comprehensive in vitro evaluation of compound degradation across a wide range of physiological and biorelevant media. Chemical stability is essential for predicting the behaviour of orally administered molecules, particularly during transit through the gastrointestinal tract where pH, buffer composition and food effects can significantly influence degradation rates. The assay evaluates non‑enzymatic degradation pathways, including hydrolysis, oxidation and other chemical transformations that may occur under different pH and environmental conditions.
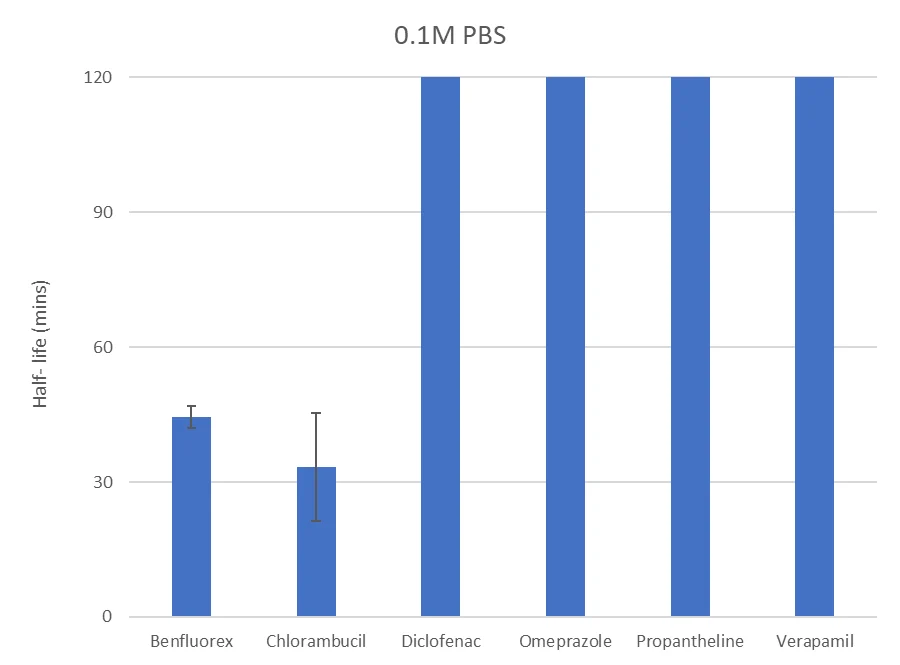
The validation assessed stability in phosphate‑buffered saline (PBS), biorelevant gastric and intestinal fluids (FaSSGF, FeSSIF, FaSSIF), fed‑state gastric media (FEDGAS) and USP‑defined buffers. Test compounds were incubated in the relevant media, and compound disappearance monitored over time using LC–MS/MS. Percent remaining and half‑life values were calculated from depletion kinetics, providing a robust measure of stability across media.
By incorporating a range of physiological and biorelevant media, the platform enables assessment of compound stability under fasted, fed, gastric, and intestinal conditions, while also supporting evaluation in formulation‑relevant environments. Overall, the dataset confirms that the chemical stability platform is an essential early‑stage DMPK tool for identifying compounds with suitable stability profiles to progress within oral drug‑development programmes.
The chemical stability assay evaluates the degradation profile of compounds across multiple physiologically relevant media to determine their intrinsic degradation rates. Test compounds are first prepared from concentrated DMSO stock solutions, then incubated in the selected buffer or biorelevant medium. At predetermined time points, aliquots are removed and quenched immediately using organic solvent to prevent further degradation, generating a time course of compound disappearance.
Following centrifugation to remove precipitated proteins, supernatants are pooled for cassette analysis (except if screening for prodrugs) and diluted with water containing internal standard. Samples are analysed via LC–MS/MS to determine the percentage parent compound remaining at each time point. Natural log transformation of compound response–time data enables linear regression to derive the elimination rate constant (k), from which half‑life values are calculated, thus providing a quantitative measure of stability.
This workflow supports high reproducibility by using consistent incubation formats, sampling schedules and analytical conditions. Overall, the assay enables efficient, accurate and physiologically relevant stability assessment for a broad range of drug candidates intended for oral administration.
Validation across all matrices demonstrated strong reproducibility and robust performance, enabling detailed stability characterisation across diverse pH conditions. Across buffer and biorelevant media, the assay showed acceptable reproducibility, with variability maintained below 20% CV.
The graph below presents the validated inter‑assay data for 0.1M PBS (pH 7.4):


Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.
