

Ressources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study











juillet 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

Tenez‑vous au courant des dernières mises à jour de l’entreprise, des étapes clés et des annonces importantes.
Découvrez les conférences et rencontres scientifiques à venir, où vous pourrez échanger avec notre équipe.

Chez Sygnature Discovery, nous fournissons des solutions de découverte de médicaments de classe mondiale pour accélérer la progression de vos composés, de l’idée jusqu’à la clinique.

Notre équipe de direction rassemble une diversité d’expériences et de ressources, favorisant la collaboration et l’innovation dans l’ensemble du processus de découverte de médicaments.

Explorez les opportunités de carrière chez Sygnature Discovery. Rejoignez une équipe internationale qui obtient des résultats exceptionnels grâce à la collaboration, à l’innovation et à l’intégrité.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
Chez Sygnature Discovery, nous fournissons des solutions de découverte de médicaments de classe mondiale pour accélérer la progression de vos composés, de l’idée jusqu’à la clinique.
Notre équipe de direction rassemble une diversité d’expériences et de ressources, favorisant la collaboration et l’innovation dans l’ensemble du processus de découverte de médicaments.
Explorez les opportunités de carrière chez Sygnature Discovery. Rejoignez une équipe internationale qui obtient des résultats exceptionnels grâce à la collaboration, à l’innovation et à l’intégrité.

Dr Steve St-Gallay – Chercheur principal, Chimie computationnelle
Le Saint Graal de la chimie computationnelle est de prédire avec exactitude, précision et régularité la puissance de liaison d’une molécule médicamenteuse potentielle avant sa fabrication, et malgré tous nos efforts (et les affirmations de certains groupes), cela n’est pas encore possible.
Les lois physiques fondamentales régissant l’interaction énergétique entre une molécule médicamenteuse et son partenaire dans l’organisme sont si complexes qu’il est impossible de les calculer sans plusieurs hypothèses. Par exemple, à l’échelle atomique, le monde fascinant de la physique quantique s’applique, mais même avec une puissance de calcul accrue, ces calculs ne peuvent être effectués dans un délai raisonnable ; nous nous référons donc à la physique de l’époque de Sir Isaac Newton.
Une autre hypothèse concerne la modification du désordre qui survient lorsqu’une molécule de médicament se lie à son site dans l’organisme. L’univers est régi par une augmentation du désordre, mais il arrive que la liaison d’un médicament entraîne une diminution de ce désordre. Calculer cette modification de l’ordre requiert la connaissance de toutes les configurations possibles, sur des échelles de temps pouvant atteindre plusieurs heures. Nous ne pouvons traiter que quelques microsecondes, au mieux, pour une seule molécule de médicament. Or, sachant qu’un comprimé de 500 mg contient près de 2 000 000 000 000 000 000 000 (soit deux sextillions) de molécules de paracétamol, nos hypothèses concernant cette modification de l’ordre sont probablement insuffisantes.
Cela étant dit, il apparaît qu’avec des calculs poussés, il est parfois possible de classer les molécules médicamenteuses potentielles similaires selon leur énergie d’interaction, avec une précision suffisante pour décider de synthétiser les plus prometteuses. La figure ci-dessous illustre comment la simulation informatique de l’évolution de la molécule, à la fois au sein de la protéine et en solution aqueuse, permet de classer les composés médicamenteux potentiels selon leur énergie de liaison. Le mot clé est « parfois » : dans environ un tiers des cas, les résultats sont satisfaisants, dans un autre tiers, ils sont acceptables, et dans le dernier tiers, ils sont totalement inefficaces.
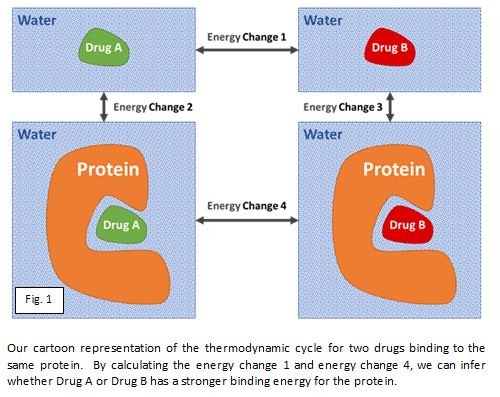
Chez Sygnature Discovery, nous avons entamé une collaboration avec un expert universitaire de premier plan dans ce domaine afin d’explorer les explications possibles quant aux cas où cette approche est précise et à ceux où elle échoue, afin de pouvoir ensuite développer des solutions informatiques pour améliorer sa précision.
L’aptitude de ces techniques à prédire les possibilités de changement de structure est particulièrement intéressante, car elle permet souvent des avancées significatives dans l’avancement des projets, moyennant un investissement plus important en chimie de synthèse. Ceci favorisera la découverte et l’optimisation de médicaments en améliorant la qualité des composés synthétisés et en réduisant les délais et les coûts.
La quête du Graal de la chimie computationnelle se poursuit, et peut-être nous en rapprochons-nous grâce à ces techniques et à une meilleure compréhension des systèmes sur lesquels elles auront le plus d’impact.
Si vous souhaitez discuter de la manière dont Sygnature Discovery peut vous aider en matière de ressources et d’expertise dans la découverte de médicaments, veuillez utiliser nos formulaires « Contactez-nous » ou nous envoyer un courriel à [email protected]






Test