

Featured Resources
-
STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months

STORM Therapeutics: From Lead to Pre-Candidate Nomination in 18 Months
Case study
📣From lead to pre-candidate nomination in 18 months. Explore the STORM Therapeutics case study







Discover precise insights into brain neurochemistry with Sygnature Discovery's in vivo microdialysis and cOFM services. With over 20 years of expertise, we design bespoke studies that reveal how compounds modulate neurotransmitter systems in health and disease. Using UHPLC/HPLC with electrochemical detection or mass spectrometry, we deliver robust PK/PD data to support confident CNS decision making.




July 30, 2015

Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.

At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.

Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.

Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Inobrodib, an exciting, first-in-class oral anti-cancer drug in clinical development by CellCentric, was collaboratively designed, synthesised and supported on its pre-clinical journey by an integrated project team at Sygnature Discovery. Inobrodib is now showing promising results in Phase I and II trials for multiple myeloma and other cancer types.
At Sygnature Discovery, we deliver world-leading drug discovery solutions to accelerate your compound from idea to clinic.
Our leadership team brings diverse experience and insight, driving collaboration and innovation across drug discovery.
Explore careers at Sygnature Discovery and join a global team committed to science, collaboration, and integrity.
Why model choice has become a data-driven discipline, and how we can support it with platforms such as DepMap, GTEx, and Open Targets
Why model choice has become a data-driven discipline in the age of novel drug modalities, and how we can support it with platforms such as DepMap, GTEx, and Open Targets
Drug discovery is expanding beyond traditional small molecules to now include biologics, cell and gene therapies, RNA-based approaches, and targeted protein degraders. Consequently, experimental systems used to evaluate targets face increasingly complex demands. Each modality affects biology differently, placing distinct requirements on the cell line models chosen to represent disease. In this context, model selection has shifted from a largely practical decision, driven by precedent, cost, or availability, to a strategic one, grounded in evidence about whether a given system reflects the biology a programme intends to modulate.
Bioinformatics is the discipline that makes this evidence accessible. By integrating functional genomics, transcriptomic, proteomic, and clinical datasets, it allows discovery teams to characterise disease-relevant biology independently of any single experimental system. We can then ask a sharper question: which available models, alone or in combination, genuinely recapitulate that biology. Here we set out the rationale for bioinformatics-led model prioritisation and describes, at the level of method, how public platforms such as DepMap, GTEx, and Open Targets are used in practice to support that process.
Cell line models fail when they do not capture the biology that matters for a given target or modality. Historically, model choice was often driven by accessibility and familiarity, rather than by direct evidence of biological relevance. The diversity of modalities now active in discovery pipelines makes that approach increasingly risky, because a model well suited to evaluating a small-molecule mechanism may be poorly suited to assessing an antibody–antigen interaction, a degrader, or a cell-based therapy.
A more principled approach starts with the disease biology itself. Genomic and transcriptomic data define which cell types express a target of interest, which pathways are dysregulated in disease, and which compensatory mechanisms might blunt a therapeutic effect. Functional genomics data add a further, causal layer: rather than only describing correlation between a gene and a disease state, large-scale loss-of-function screens reveal whether a gene is actually required for the survival or proliferation of a given cell type1. Together, these layers let teams characterise the biological landscape before committing to a specific model system, rather than retrofitting biological rationale onto whichever model is already on hand.
Mapping the Dependency Landscape
A natural starting point for model prioritisation is the dependency landscape of a target across cancer cell line models. The Cancer Dependency Map (DepMap) project provides genome-scale CRISPR and RNAi loss-of-function screening data across more than a thousand cancer cell lines, expressed as a gene-effect or “dependency” score. A strongly negative score indicates that silencing or deleting a gene impairs cell survival or growth in that line, analogous to a gene’s essentiality, and so flags a genetic vulnerability that may be therapeutically exploitable.
Plotting dependency scores by lineage (Figure 1), reveals which cancer types are most reliant on a given target and which are not. Conventional thresholds are used to interpret the score: a mean dependency at or below –0.5 is generally considered evidence that a gene is essential in that lineage, while a score at or below –1 indicates strong essentiality, comparable to the median effect of genes classed as commonly essential across the entire cell line panel. Analysing at essentiality scores gives an immediate, data-driven shortlist of which cell line models are mechanistically appropriate for a target evaluation programme, rather than relying on assumptions carried over from prior projects. For example, here we can see that all cell lines in the Eye cancer lineage (pink) show a dependency on MDM2.
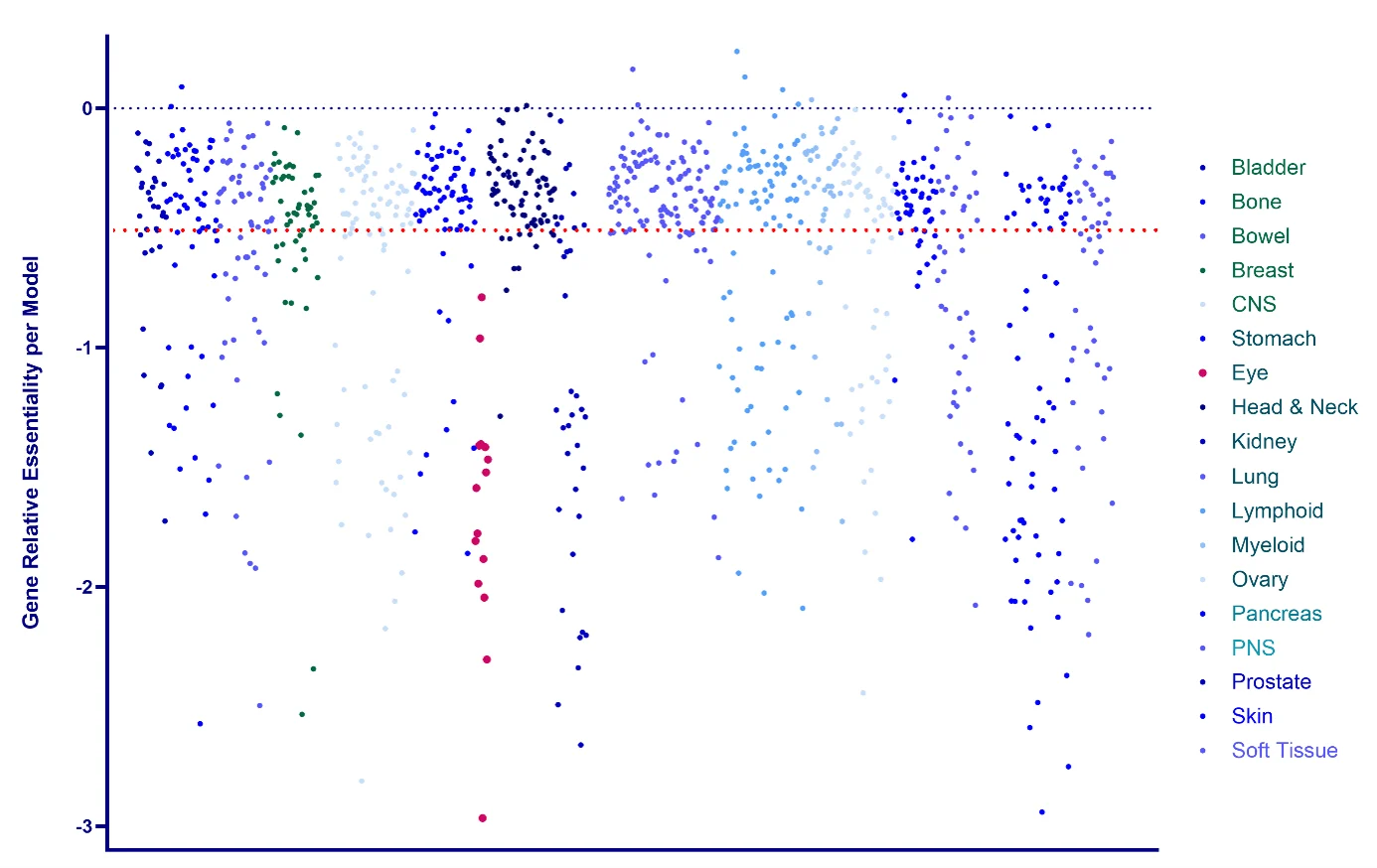
Figure 1. MDM2 ‘Essentiality’ across 18 Oncology lineages. Individual cell lines, group by lineage, MDM2 essentiality. ≤-0.5: Standard threshold (red line) indicating that a gene is considered essential in that specific cancer cell line. ≤-1: Indicates a strongly essential gene (equivalent to the median effect of all “common essential” genes).
Integrating Expression with Dependency
Dependency alone does not give the full picture, however. Expression of a target is a useful complementary signal, and one that has been widely used in target identification. Importantly, its relevance varies by modality and expression-driven rationale tends to carry more weight for small molecules acting on enzymatic or signalling activity than it does for some other mechanisms. Combining CRISPR or RNAi dependency data with expression data, drawn from resources such as cBioPortal or GTEx, builds a stronger and more falsifiable target rationale than either dataset alone.
Correlating dependency and expression across a panel of models, typically shows that higher expression of a target tracks with greater essentiality across most lineages. The exceptions are informative in their own right: a lineage that shows strong essentiality without a corresponding expression correlation suggests the dependency is being driven by a mechanism other than simple target abundance, which is a meaningful flag for follow-up mechanistic work rather than a result to be discarded.
This kind of correlation analysis is also a practical input to modality selection. Where dependency and expression are tightly coupled across a large panel of models, and where orthogonal screening methods such as CRISPR knockout and RNAi knockdown agree, confidence in the underlying biology is higher and the choice between modalities, for example, an antibody drug conjugate that depends on both target expression and payload-driven killing, can be made on firmer ground.
Dependency and expression data become considerably more actionable once stratified by biomarkers like a genomic subtype. Enrichment of a dependency signal by lineage or molecular subtype, and its association with recurrent mutations, for example in TP53, BRCA, or KRAS, can encompass structural variants, damaging or hotspot mutations, variant allele frequency, and copy-number alteration alongside expression, network correlation, and clinical metrics.
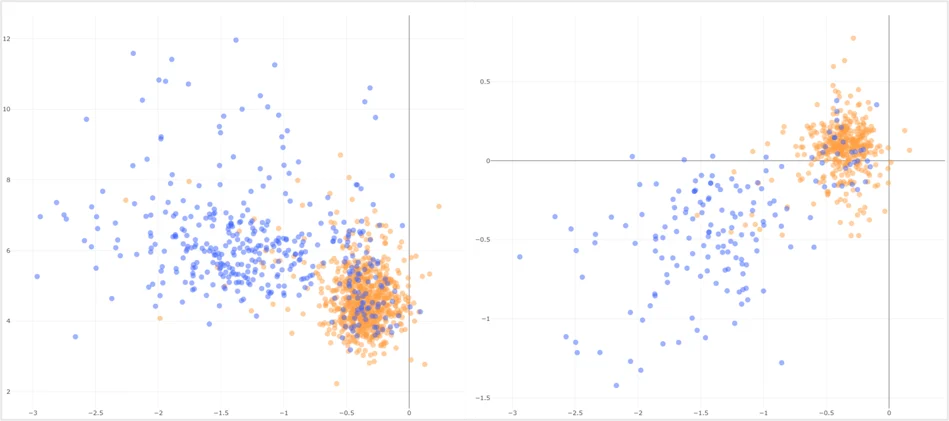
Figure 2. MDM2 Essentiality correlates with elevated MDM2 expression in >1,100 oncology models (left). Significantly, wild-type p53 (Blue) is a clear marker of MDM2 essentiality in the majority of models, highlighting a biomarker to define MDM2-targetable vulnerability. Importantly, in the right panel we can see that RNAi and CRISPR-KO screening show strong correlation for MDM2 dependency. This data is critical to support drug modality selection.
Across large panels of models (Figure 2) can surface a clear biomarker defining a vulnerability for instance, a particular mutational status segregating cleanly with essentiality across the majority of models. Extending this comparison from cell line panels into patient cohort data (Figure 3), strengthens the rationale further. Where mutations in a target gene co-occur with other prognostically significant mutations in pan-cancer patient data, that pattern helps define a clinically relevant, biomarker-defined population for therapeutic intervention. Ultimately this de-risks the target by anchoring cell-line-derived hypotheses to evidence from patients.
Assessing Risk and Selectivity
A target that is essential in disease-relevant models is not automatically a good target. In fact, selectivity matters as much as dependency. Comparing dependency across the full panel of lineages makes it possible to distinguish targets that are selectively required in disease-relevant cell types from those that are broadly essential across most or all lineages, the latter carrying a substantially higher risk of on-target toxicity.

Figure 3. MDM2 mutations co-occur with prognostically significant mutations in pan-cancer patient cohorts. A majority of statistically significant mutations co-occur in MDM2 mutant patients (left-panel; black) while MDM2 wild-type patients show a significant co-occurrence with p53 mutations (left-panel, red). Indeed, >40% of patients with wild-type MDM2 carry a p53 mutation, while >40% of patients with MDM2 mutations also carry a CDK4 mutation (right-panel).
Healthy-tissue expression data add a necessary safety dimension to this picture. Bulk RNA-seq atlases such as GTEx, and single-cell expression resources, show how a target is expressed across normal tissues (Figure 4). Bulk data can flag tissues with disproportionately high expression, while single-cell data adds resolution that bulk profiling cannot. A target might appear only moderately expressed at the tissue level while in fact being highly expressed in a discrete subpopulation of cells within that tissue, a pattern that bulk RNA-seq alone would mask entirely. Folding this kind of healthy-tissue triage, alongside known toxicity issues, prior clinical trial failures (and potentially freedom-to-operate considerations) into a structured deprioritisation score allows target and model long-lists to be triaged.
From Single Models to Coherent Model Portfolios
For complex programmes, no single model or single dataset is sufficient. Confidence accumulates from concordance across complementary systems and data types: dependency screens, expression atlases, biomarker and patient cohort data, and increasingly, complex human cell models such as organoids, primary cell cultures, and advanced three-dimensional systems. These models capture aspects of tissue architecture and cellular heterogeneity that two-dimensional cultures cannot. However, their added complexity also raises the bar for characterisation. Critically though, this increased biological richness only translates into better decisions when it is benchmarked rigorously against clinically relevant data.
Integrative analysis allows signals from different models (e.g. 2D cell systems, organoids, and in vivo models) to be evaluated against a shared biological framework. The relevant question is rarely whether models agree in absolute, quantitative terms, but rather whether they converge on the same pathways, functional outcomes, or resistance mechanisms. That convergence is often more informative than numerical alignment, and bioinformatics is the connective layer that makes it visible, turning a set of disparate experimental systems into a coherent biological narrative2,3.
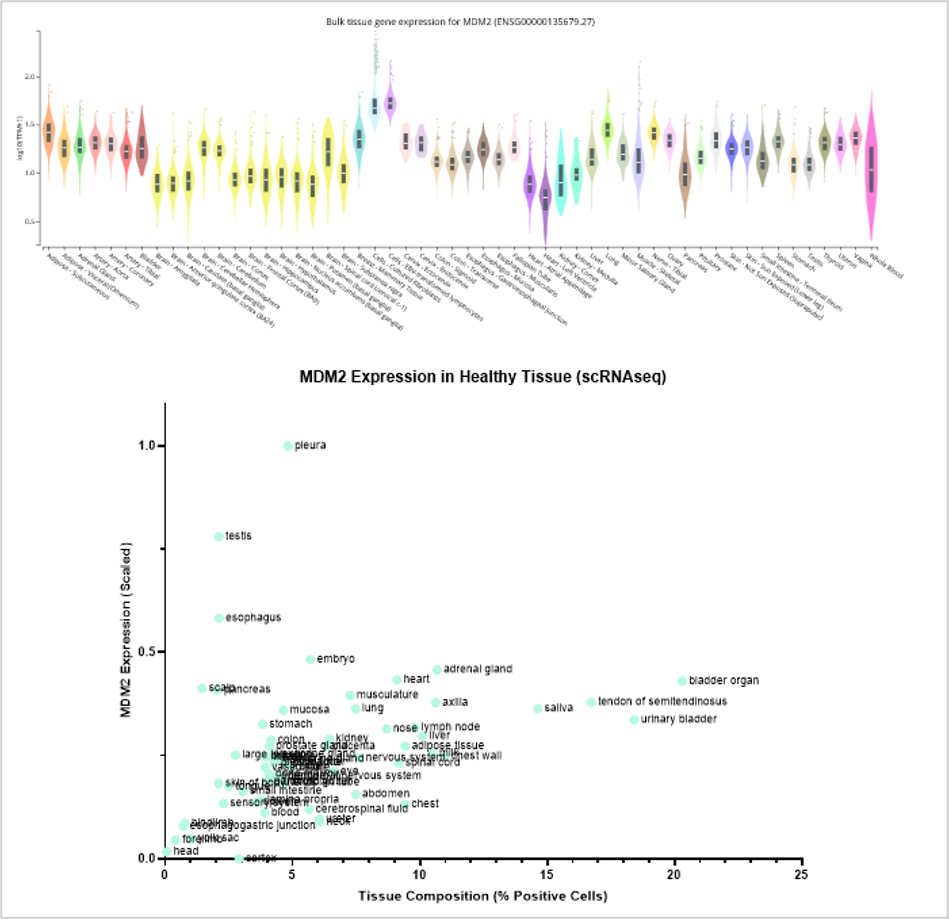
Figure 4. MDM2 Expression in healthy tissue. MDM2 is highly expressed in lung tissue and adipocytes as determined by bulk RNAseq (top-panel). MDM2 is also shows as highly expressed in ~5% of pleura cells, but moderately expressed in a broad range of tissues (e.g. >20% of bladder organ cells) as determined by single cell RNAseq (lower-panel).
Where Open Targets Fits
Open Targets sits alongside DepMap and GTEx as a third pillar of this workflow. The platform aggregates genetic association, expression, pathway, and tractability evidence to support target identification and prioritisation at the disease level. Where DepMap addresses functional dependency in cancer models and GTEx addresses normal-tissue expression context, Open Targets contributes a broader evidence base linking targets to disease through human genetics and prior drug programme outcomes. Used together, these platforms let a discovery team move from “which models are dependent on this target” to “does the genetic and clinical evidence support this target in this disease” to “is this target likely to be safe and modality-appropriate” within a single, evidence-linked workflow, rather than treating each question in isolation.
Implications for Drug Modality Selection
Different modalities place different demands on the evidence generated through this process. Small molecules typically depend most heavily on faithful recapitulation of enzymatic activity or signalling dynamics, making dependency and pathway-level data especially informative. Biologics depend more on cellular context, receptor architecture, and species-specific interactions, which raises the importance of expression localisation and cross-species comparison. Cell and gene therapies introduce further considerations around differentiation state, tissue microenvironment, and long-term functional stability, which complex human cell models are increasingly used to address.
Framing model selection as a data-driven matching exercise between modality mechanism and model biology, rather than a binary or default choice, lets bioinformatics surface modality-specific risk earlier in a programme. Expression analyses can show whether a target’s cellular localisation is compatible with antibody engagement, whether an RNA-based therapeutic is likely to reach disease-relevant compartments, or whether pathway redundancy may blunt the effect of targeted protein degradation. These insights do not replace experimental work. Instead they focus it on the models and assays most likely to expose a genuine limitation. This reduces the chance that a programme carries a hidden biological assumption downstream into more costly stages of development.
Further Implications and Utility of DepMap Data
Beyond the essentiality of any single gene, DepMap supports co-dependency analysis: identifying other genes whose dependency profiles correlate strongly, whether positively or negatively, with a gene of interest across the full cell line panel4. Genes that cluster together by dependency pattern are often functionally connected, members of the same complex, pathway, or process, even where no prior biochemical link is known. This effectively turns DepMap into a tool for inferring gene function and pathway membership from phenotypic data alone, independent of expression or sequence homology.
A practical consequence of this is target hopping: where a gene of interest is biologically validated but poorly druggable, co-dependency clustering can surface alternative, more tractable genes that share a similar selectivity profile and are likely to act through the same pathway. This reframes model and target prioritisation as a network-level exercise rather than a one-gene-at-a-time search and is increasingly used to recover programmes that would otherwise stall on an undruggable node.
Synthetic Lethality and Paralog Buffering
Single-gene dependency screens can underestimate true vulnerability where a gene’s function is buffered by a paralog — a second gene, arising from earlier duplication, that can partially compensate when the first is lost. Loss of either gene alone may be tolerated, while combined loss is lethal. Systematic analysis of paralog pairs against DepMap dependency data, integrated with expression and mutation context, has been used to predict thousands of such context-dependent synthetic lethal relationships across large cell line panels, most of which would be invisible to single-gene knockout screening alone.
This matters directly for cell model prioritisation: a model selected purely on single-gene essentiality may appear to show no dependency on a biologically important gene, simply because a paralog is compensating in that particular line. Recent work has also used DepMap screening data to uncover synthetic lethal relationships driven by recurrent copy-number loss rather than paralogy, for example, a gene dependency restricted to cell lines carrying a specific co-deleted chromosomal region, identifying biomarker-defined patient populations for genes with no previously known drug. Building paralog and copy-number context into model selection therefore reduces the risk of discarding a genuinely valid target because the wrong cell lines were tested.
Linking Genetic Dependency to Drug Sensitivity
DepMap’s PRISM repurposing dataset profiles thousands of small molecules, including many non-oncology agents, across hundreds of cell lines, generating a pharmacological counterpart to the genetic dependency data. Comparing a gene’s CRISPR dependency profile against the sensitivity profile of drugs annotated to that same target provides an orthogonal check on target validity: where genetic loss-of-function and pharmacological inhibition produce correlated selectivity across the same lines, confidence that the drug acts through the expected mechanism is considerably higher.
This comparison also has direct value for repurposing and unexpected-mechanism discovery: selective cytotoxicity has been linked back to gene dependencies for agents not originally developed as cancer therapeutics, where sensitivity tracked with a specific gene’s expression or dependency rather than the drug’s nominal target. For model selection specifically, this means a candidate cell line panel can be screened not only for genetic dependency on a target, but for differential sensitivity to existing tool compounds or clinical-stage agents, adding a pharmacological layer of evidence before committal to a full assay cascade.
A Methodological Caveat: Copy-Number Artefacts in Dependency Scores
Raw CRISPR dependency data carry a known artefact worth understanding before scores are used to rank or shortlist models: Cas9-mediated DNA cleavage itself produces a gene-independent anti-proliferative effect in regions of high copy-number amplification, simply because more cut sites are being introduced, independent of whether the targeted gene matters biologically. Left uncorrected, this makes genes in amplified regions appear more essential than they actually are, which is a particular risk in cancer types with extensive genomic instability.
DepMap addresses this through computational correction models, first CERES, and more recently Chronos, that explicitly separate the copy-number-driven cutting effect from true gene-knockout effect when computing the gene-effect score. The dependency scores referenced earlier in this note are the corrected output of this process, not raw screen depletion values. This is worth flagging explicitly because it affects how dependency data should be interpreted in practice: a strong dependency signal in a heavily amplified region still warrants a sanity check against orthogonal evidence (RNAi screening data, expression, or PRISM drug sensitivity) before being treated as a confirmed vulnerability, and the corrected, rather than raw, dependency values should always be the basis for model shortlisting.
As drug modalities diversify, the value of bioinformatics lies less in generating predictions than in structuring the thinking behind model choice. This is reflected in whether a candidate model is appropriate for the relevant disease biology, or whether it is appropriate for a given modality’s mechanism of action. Similarly, we can ask whether the available evidence, from dependency screens, expression atlases, biomarker data, and disease-level platforms such as Open Targets, converges to support advancing a target. The most effective discovery teams are not necessarily those using the most sophisticated individual model. The most effective are using bioinformatics to connect dependency, expression, biomarker, and risk data into a single, evidence-linked strategy for cell line model prioritisation. As therapeutic complexity continues to increase, that capability is becoming foundational to discovery rather than optional.
Contact us to discuss how data-driven approaches and leading bioinformatics resources can accelerate your drug discovery programs.
References


Peak Proteins, NuChem Sciences, and SB Drug Discovery have now fully integrated with Sygnature Discovery.
